Greig-Syndrom
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.0 | Angeborene Fehlbildungssyndrome mit vorwiegender Beteiligung des Gesichtes |
| ICD-10 online (WHO-Version 2019) | |

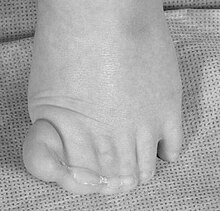
Das Greig-Syndrom ist eine angeborene Erkrankung mit einer charakteristischen Kombination von Schädel-Gesichtsdysmorphie und Polydaktylie (Vielgliedrigkeit der Finger).[1][2]
Synonyme sind: Greig-Zephalopolysyndaktylie-Syndrom; Zephalopolysyndaktylie; Hootnick-Holmes-Syndrom
Nicht zu verwechseln ist der Familiäre Hypertelorismus Greig.[3][1]
Die Bezeichnungen beziehen sich auf den Autor der Erstbeschreibung 1926 durch den schottischen Arzt David Middleton Greig (1864–1936)[4][5] sowie die Autoren einer Publikation von 1972, den US-amerikanischen Orthopäden David Randall Hootnick und den US-amerikanischen Kinderarzt Levis B. Holmes.[6][7]
Vorkommen
Die Häufigkeit wird mit 1–9 zu 1.000.000 angegeben, die Vererbung erfolgt autosomal-dominant.[2]
Ursache
Der Erkrankung liegen ursächlich Loss-of-Function-Mutationen im GLI3-Gen auf Chromosom 7 am Genort p13 zugrunde, welches für einen Transkriptionsfaktor kodiert.[8]
Andere Mutationen im gleichen Gen finden sich bei dem Pallister-Hall-Syndrom und dem Akrokallosalen Syndrom.[2]
Klinische Erscheinungen
Diagnostische Kriterien sind:[1][2]
- Makrozephalus, prominente Stirn, später Fontanellenschluss
- Hypertelorismus
- Polydaktylie der Hände (postaxial) und der Füße (präaxial)
- Partielle häutige Syndaktylien der Finger & Zehen II-V
selten zusätzlich Balkenmangel, andere Anomalien des Gehirnes.
Diagnostik
An das Syndrom sollte beim Vorliegen der „klassischen“ Trias von präaxialer Polydaktylie mit häutiger Syndaktylie, Hypertelorismus und Makrozephalie gedacht werden.[2]
Differentialdiagnose
- Akrokallosales Syndrom
- Carpenter-Syndrom
- Gorlin-Goltz-Syndrom
- Hypertelorismus vom Typ Teebi
- Mohr-Syndrom
- Pallister-Hall-Syndrom
- Polysyndaktylie
Heilungsaussicht
Es besteht ein etwas höheres Risiko einer auch geistig verzögerten Entwicklung.[2]
Literatur
- J. Mücke, K. R. Sandig: Greig-Syndrom. In: Kinderärztliche Praxis, Februar 1985, Band 53, Nr. 2, S. 89–91; ISSN 0023-1495. PMID 3921750.
- L. G. Biesecker: The Greig cephalopolysyndactyly syndrome. In: Orphanet Journal of Rare Diseases, 2008, Band 3, S. 10; ISSN 1750-1172. doi:10.1186/1750-1172-3-10. PMID 18435847. PMC 2397380 (freier Volltext). (Review).
Weblinks
- L. G. Biesecker, J. J. Johnston: Greig Cephalopolysyndactyly Syndrome. In: GeneReviews®
- Medline Plus
Einzelnachweise
- ↑ a b c d Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- ↑ a b c d e f g Greig-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- ↑ D. M. Greig: Hypertelorism. A hitherto undifferentiated congenital cranio-facial deformity. In: Edinburgh Medical Journal, 1924, Band 31, S. 560–593.
- ↑ Who named it Greig
- ↑ D. M. Greig: Oxycephaly In: Edinburgh Medical Journal, 1926, Band 33, S. 189–218.
- ↑ D. Hootnick, L. B. Holmes: Familial polysyndactyly and craniofacial anomalies. In: Clinical Genetics, Copenhagen, 1972, Band 3, S. 128–134.
- ↑ Greig's Syndrom. Who named it.
- ↑ Greig cephalopolysyndactyly syndrome. In: Online Mendelian Inheritance in Man. (englisch)

Dieser Artikel behandelt ein Gesundheitsthema. Er dient nicht der Selbstdiagnose und ersetzt nicht eine Diagnose durch einen Arzt. Bitte hierzu den Hinweis zu Gesundheitsthemen beachten!








